O QUE É IMPORTANTE CONHECER?
A denominação remodelamento foi utilizada inicialmente para descrever os câmbios produzidos após o infarto do miocárdio, particularmente a dilatação do VE (1). Hoje sabemos que o remodelamento cardíaco descreve várias situações clínicas e mudanças fisiopatológicas, determinando vários fenótipos diferentes, conforme o tipo de injúria e a cavidade ou cavidades afetadas.
Chama-se remodelamento a ocorrência de alterações moleculares, celulares e intersticiais que se manifestam como mudanças no tamanho, forma e função do coração após uma lesão cardíaca, sobrecarga pressórica ou volumétrica ou, ainda, devido a uma adaptação fisiológica (2).
O remodelamento do VE provoca mudanças no diâmetro, massa (hipertrofia ou atrofia), geometria (espessura das paredes e forma da cavidade), áreas de cicatriz após infarto do miocárdio, fibrose ou infiltrado inflamatório (miocardites). Os métodos utilizados paras detectar essas mudanças são a ecocardiografia, a ventriculografia e a ressonância magnética cardíaca. Outras formas de diagnóstico, ainda não utilizadas na rotina clínica, são a detecção de marcadores celulares que manifestam a mudança de expressão de alguns genes que modificam as cadeias da proteínas contráteis, alterando as condições bioquímicas celulares (3).
A principal consequência do remodelamento patológico é a disfunção miocárdica, marcando o início e a progressão da disfunção ventricular, que começa com mudanças genéticas em resposta ao dano cardíaco, provocando alterações celulares e moleculares que levam à perda progressiva da função ventricular, assintomática primeiro, evoluindo depois para sintomas e sinais de insuficiência cardíaca.
Os eventos começam com o dano miocárdico, que pode ser causado por apoptose de miócitos (como ocorre no infarto do miocárdio), por inflamação (como ocorre nas miocardites), por causas tóxicas (como acontece na toxicidade por agentes quimioterápicos), por mudanças genéticas (como visto nas cardiomiopatias por mutação de proteínas sarcoméricas), por sobrecargas pressóricas volumétricas (como ocorre na hipertensão e nas valvopatias).
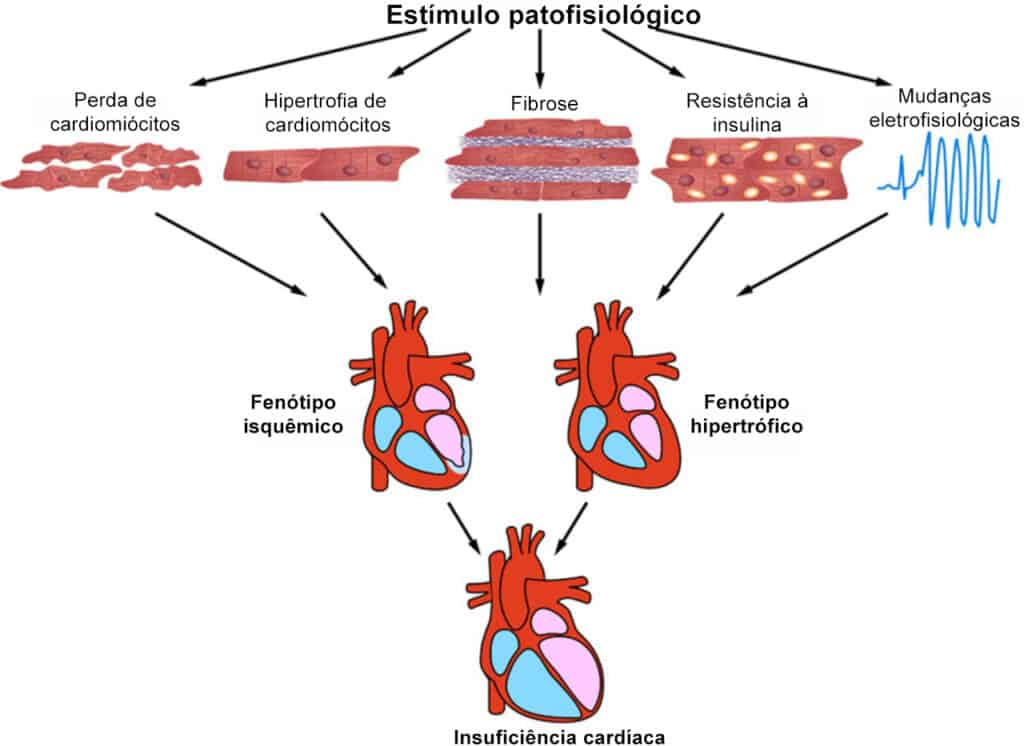
O remodelamento miocárdico pode-se associar com arritmias malignas, como taquicardia ventricular sustentada e fibrilação ventricular, sendo suas principais causas alterações nos canais iônicos (inativação dos canais de sódio, alterações nos canais de cálcio e potássio e mudanças no intercâmbio sódio/cálcio). Outras mudanças ocorrem nos discos intercalados com alteração das conexões intercelulares, provocando aumento do intervalo QT e arritmias. O aumento do colágeno, nos três compartimentos miocárdicos (epimísio, que envolve todas as fibras musculares, perimísio, que envolve feixes menores de miócitos e endomísio, que separa os próprios miócitos) provocando fibrose, bloqueia os estímulos elétricos, diminui a velocidade de transmissão dos impulsos, aumenta o tempo de período refratário e pode promover fenômenos de reentrada que provocam arritmias, cuja principal consequência é a morte súbita.

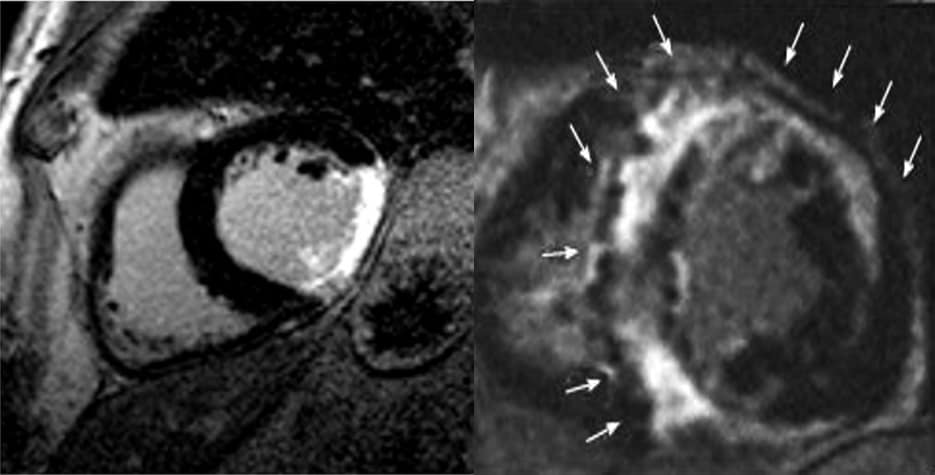
Como a morte súbita é a principal causa de morte após o infarto do miocárdio (em indivíduos mais idosos) e nas cardiomiopatias genéticas (indivíduos jovens), a pesquisa e identificação da fibrose miocárdica adquire grande importância. A ressonância magnética cardíaca mostra claramente essas áreas de fibrose.
Com a ecocardiografia pode-se ser determinada a denominada heterogeneidade elétrica, provocada pelas áreas de fibrose. Esta heterogeneidade elétrica, devida à alteração da velocidade da transmissão do estímulo provoca, no ECG de alta resolução, alongamento regional do intervalo QT e pode ser detectado pela ecocardiografia com strain longitudinal, pela variação do tempo de deformação de cada segmento miocárdico. Essa técnica é denominada “dispersão mecânica”. O tempo ao pico da máxima deformação também se associa com a ocorrência de arritmias (4). Atualmente, considera-se dispersão mecânica aumentada com maior probabilidade de arritmias um desvio-padrão do tempo ao pico >61 ms.

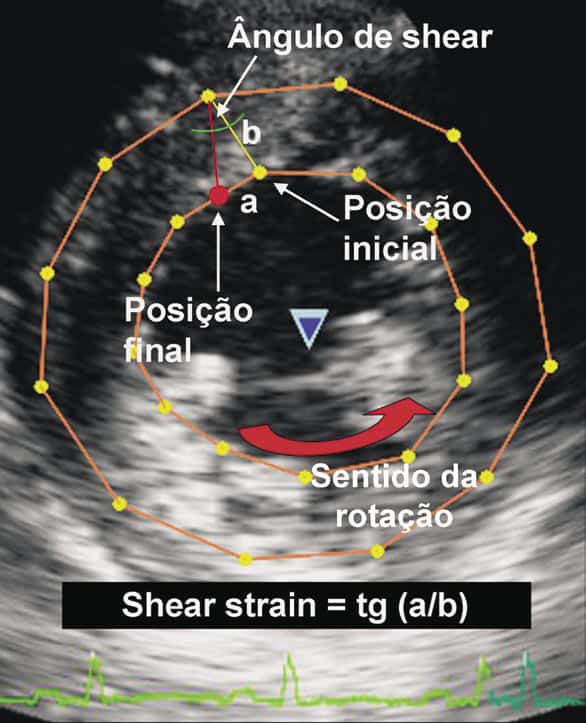
O infarto do miocárdico provoca, nas primeiras horas de sua ocorrência, desintegração do colágeno interfibrilar (com a consequente perda da matriz intersticial), juntamente com necrose ou miofibrólise celular. Esta perda do tecido de sustentação faz esta área susceptível de distensão e deformação, que se traduz por adelgaçamento da região infartada e dilatação da cavidade, como consequência do deslizamento das células necróticas e reacomodação dos miócitos ao longo da parede infartada. A dilatação ventricular aguda, caracterizada por afilamento e alongamento do infarto, que é chamada expansão do infarto, aumenta o potencial de ruptura miocárdica e forma o substrato para os aneurismas (1). Já, a forma crônica, é responsável pelos aneurismas pós-infarto. Novas técnicas estão em desenvolvimento para prever os aneurismas do VE pós-infarto, tanto pela ressonância magnética como pela ecocardiografia de strain miocárdico, analisando a deformação por cisalhamento, conhecida como “shear strain”. Como a perda da matriz intersticial provocada pelo remodelamento pós-infarto provoca maior deslizamento das camadas que formam a parede do miocárdio, a deformação por cisalhamento aumenta, podendo ser usada como marcador de remodelamento adverso (aneurisma) (5,6).
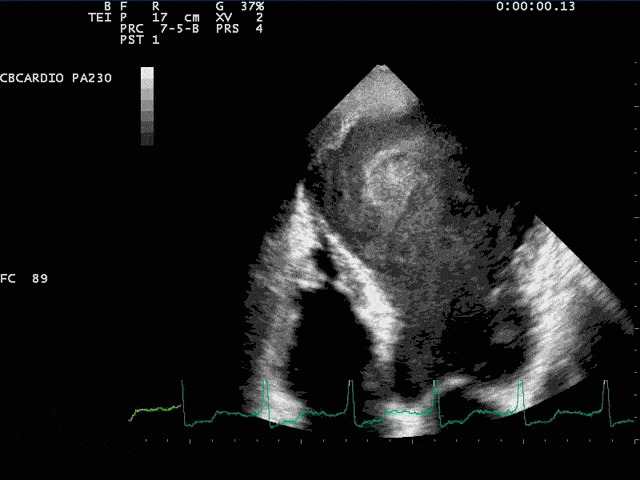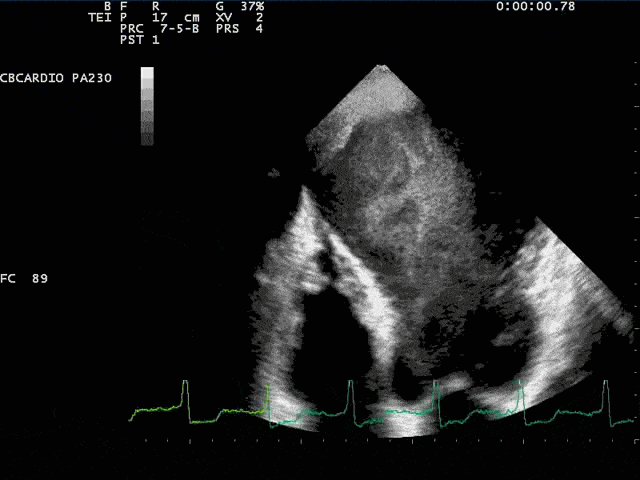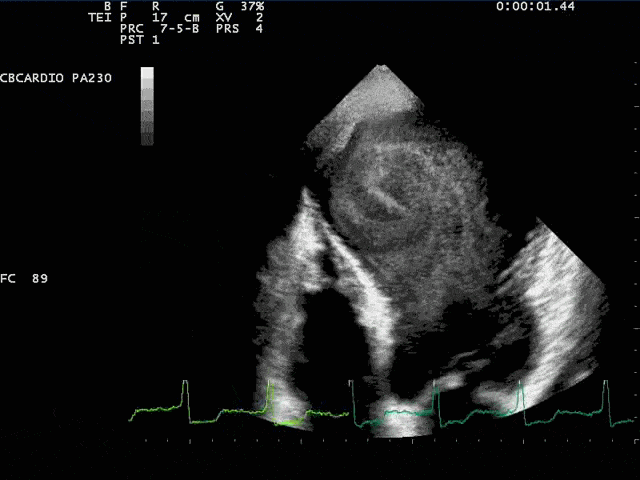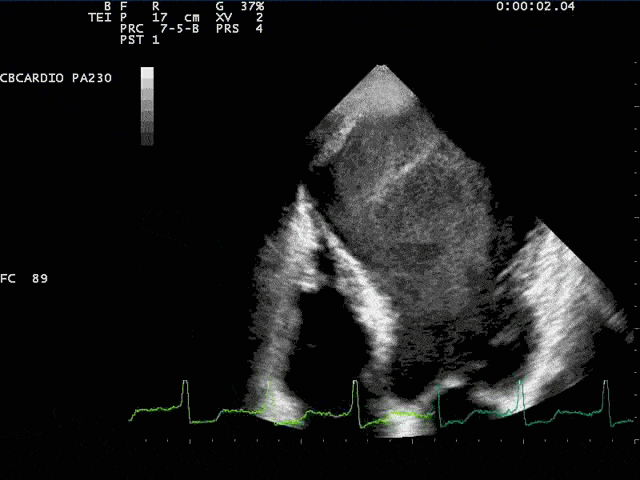
Os mecanismos da disfunção miocárdica começam com a morte celular (que pode ser provocada por apoptose, por morte programada, por necrose ou por autofagia, processos estes denominados “necroptose” (7)). A diminuição do suprimento de oxigênio diminui a oxidação dos ácidos graxos (principal componente energético do miocárdio) aumentando a oxidação da glicose, com alteração da função mitocondrial, diminuído a energia disponível para as proteínas miocárdicas provocando a geração de espécies reativas de oxigênio, causando estresse oxidativo. Estresse oxidativo ocorre quando se gera excesso de espécies reativas de oxigênio não devidamente neutralizadas pelos sistemas antioxidantes (8). Os mecanismos autoimunes produzem um aumento das citosinas (resposta inata) e um processo inflamatório mais específico (resposta adaptativa), mediado por macrófagos e células regulatórias B e T, que parecem ser benéficas, produzindo um processo de remodelamento mais favorável em modelos de isquemia (9).
O interstício é formado por 95% de fibras de colágeno tipo I e tipo III, responsáveis pela regulação da apoptose, pela reparação de deformações patológicas, pela manutenção do alinhamento das estruturas, pela regulação da distensibilidade (complacência) do músculo cardíaco e pela transmissão do esforço durante a contração muscular. As fibras de colágeno estão unidas por ligações químicas resistentes à degradação da maioria das proteases, mas algumas enzimas, como a metaloproteinase, tem atividade colagenolítica. A ruptura da rede de colágeno, com perda da matriz intersticial, leva a alterações da arquitetura e função ventriculares nos modelos de infarto agudo. Por outro lado, o acúmulo de colágeno tipo III e, principalmente tipo I (mais rígido, com fibras mais longas e mais estável) observados em outros tipos de injúria cardíaca aumenta a fibrose com aumento da rigidez do miocárdio, disfunção diastólica, diminuição da contratilidade, diminuição do fluxo coronariano e ocorrência de arritmias malignas, sendo a fibrose importante preditor de mortalidade (10).
O remodelamento miocárdico também modifica uma das principais proteínas contráteis, a miosina, composta por dois pares de cadeias pesadas e por dois pares de cadeias leves que, ao se alterar diminuem a contratilidade. No remodelamento o transporte do cálcio através do retículo sarcoplasmático (canais de cálcio) encontra-se reduzido durante a sístole e aumentado durante a diástole. Como consequência temos mudanças da geometria (de forma elíptica para esférica) e da espessura das paredes, com diminuição da contratilidade e da rotação e torção miocárdicas e perda de eficiência mecânica sistólica (nos modelos de isquemia) e diastólica (em todas as formas de injúria). Com relação à ativação neuro-hormonal, no remodelamento estão envolvidos o sistema simpático e o sistema renina-angiotensina-aldosterona. A ativação de ambos os sistemas estimula a síntese de proteínas em miócitos e fibroblastos causando hipertrofia ventricular e fibrose. Outros efeitos incluem ativação de fatores de crescimento e metaloproteinase, sobrecarga hemodinâmica por vasoconstrição e retenção de água, aumento do estresse oxidativo e efeito citotóxico direto, seguido de morte celular por apoptose ou necrose (11).
As consequências resultam, em primeiro lugar, em disfunção cardíaca subclínica, que pode ser detectada pela diminuição do strain longitudinal global e pela alteração da função diastólica; segue a fase de disfunção clínica, com diminuição da fração de ejeção, dilatação da cavidade e adelgaçamento das paredes (nas formas dilatadas) ou espessamento das paredes com relativa manutenção da fração de ejeção (nas formas hipertróficas). A agressão direta aos miócitos (cardiotoxicidade, miocardites) provoca diminuição da contratilidade com pouco aumento dos diâmetros ventriculares e alterações na função diastólica.
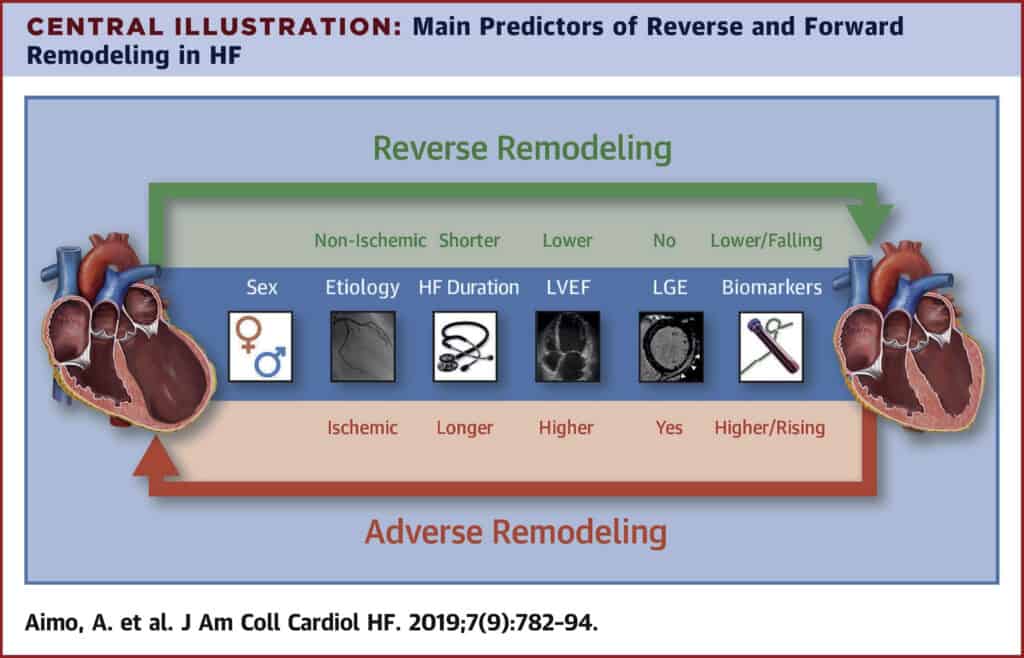
A identificação de preditores confiáveis de remodelamento permite adotar uma abordagem terapêutica adequada para cada tipo de injúria, um seguimento mais detalhado e uma planificação mais eficiente da terapia para pacientes com maior risco de dilatação progressiva do VE e declínio da fração de ejeção e, vice-versa, para aqueles pacientes que apresentam menor risco (12).
Referências.
- Pfeiffer MA, Braunwald E. Ventricular remodeling after myocardial infarction: experimental observations and clinical implications. Circulation 1990; 81(4):1161.
- Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling – Concepts and clinical implications: a consensus paper from an International Forum of Cardiac Remodeling. J Am Coll Cardiol 2000; 35:569.
- Expert Group on Biomarkers. Biomarkers in cardiology – Part 1 – In heart failure and specific cardiomyopathies. Arq Bras Cardiol 2014; 103(6):451.
- Castillo JMD, Silveira CAM, Albuquerque ES et al. Dispersão mecânica. Arq Bras Cardiol Imagem Cardiovasc 2014; 27:197.
- Castillo JMD, Herzkowicz N, Boschilia T et al. Deformação miocárdica tangencial (shear strain) em indivíduos normais: O seu significado. Rev Bras Ecocardiogr Imagem Cardiovasc 2009; 22:20.
- Gotte MJW, Germans T, Russel IK et al. Myocardial strain and torsion quantified by cardiovascular magnetic resonance tissue tagging: Studies in normal and impaired left ventricular function. J Am Coll Cardiol 2006; 48:2002.
- Xie M, Burchfield JS, Hill JA. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013; 128(4):388.
- Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 2005; 115:500.
- Mann DL. Innate immunity and the failing heart. The cytokine hypothesis revisited. Circ Res 2015; 116:1254.
- Leask A. Getting to the heart of the matter. New insights into cardiac fibrosis. Circ Res 2015; 116:1269.
- Florea VG, Cohn JN. The autonomic nervous system and heart failure. Circ Res 2014; 114:1804.
- Aimo A, Gaggin HK, Barison A et al. Imaging, biomarker, and clinical predictors of cardiac remodeling in heart failure with reduced ejection fraction. J Am Coll Cardiol: Heart Failure 2019; 7:782.

É doutor em medicina, especialista em Cardiologia (SBC) e especialista em Ecocardiografia.
Curriculum completo disponível na Plataforma Lattes
(http://lattes.cnpq.br/4922446519082204)
Obrigada pelos esclarecimentos e de forma muito didática
Óptimo resumo!